AREXVY
Laboratoire : GSK Vaccines
Le 26/01/2026. Autorisation européenne pour une utilisation élargie chez tous les adultes âgés de 18 ans et plus (en attente du nouveau Résumé des caractéristiques du produit).
RCP du 26/11/2025. Arexvy peut être administré de manière concomitante avec un vaccin à ARNm contre la covid 19, un vaccin pneumococcique conjugué, un vaccin contre le zona (recombinant, avec adjuvant) ou avec un vaccin inactivé contre la grippe saisonnière (dose standard sans adjuvant, haute dose sans adjuvant, ou dose standard avec adjuvant).
Le pharmacien est autorisé à prescrire et administrer ce vaccin.
Vaccination au calendrier vaccinal, recommandée en période épidémique (de septembre à janvier) chez les personnes ≥ 75 ans, ou ≥ 65 ans à risque. Le vaccin n'est pas encore remboursé dans cette indication.
Ce vaccin n'est pas autorisé chez la femme enceinte.
Description
Vaccin contre le virus respiratoire syncytial (VRS), recombinant, antigène RSVPreF3 dérivé de la protéine F, avec adjuvant.
Forme et Présentation
▼ Ce symbole indique que ce médicament fait l'objet d'une surveillance supplémentaire qui permettra l'identification rapide de nouvelles informations relatives à la sécurité. Les professionnels de la santé déclarent tout effet indésirable suspecté.
Poudre et suspension liquide à transformer en suspension injectable.
Vaccin du Virus Respiratoire Syncytial (VRS) (recombinant, avec adjuvant).
Poudre et suspension pour suspension injectable.
- La poudre est blanche.
- La suspension est un liquide opalescent, incolore à brun pâle.
Nature et contenu de l’emballage extérieur
Arexvy se présente sous la forme suivante :
- Poudre pour 1 dose en flacon (verre de type I) avec un bouchon (caoutchouc butyl) et un capuchon amovible vert moutarde (antigène).
- Suspension pour 1 dose en flacon (verre de type I) avec un bouchon (caoutchouc butyl) et un capuchon amovible brun (adjuvant).
Arexvy est disponible en boîte d'1 flacon de poudre et 1 flacon de suspension, ou en boîte de 10 flacons de poudre et 10 flacons de suspension.
Toutes les présentations peuvent ne pas être commercialisées.
Composition
1. Antigène
Après reconstitution, une dose (0,5 mL) contient :
- Antigène2,3 RSVPreF31 : 120 microgrammes
2 RSVPreF3 produite sur cellules d'Ovaires de Hamster Chinois (CHO) par la technique de l'ADN recombinant ;
3 Avec l'adjuvant AS01E contenant :
- Extrait de plante Quillaja saponaria Molina, fraction 21 (QS-21) : 25 microgramme ;
- 3-O-désacyl-4’-monophosphoryl lipide A (MPL) issu de Salmonella minnesota : 25 microgrammes.
2. Excipients
2.1. Poudre (antigène RSVPreF3)
- Tréhalose dihydraté
- Polysorbate 80 (E 433)
- Dihydrogénophosphate de potassium (E 340)
- Phosphate dipotassique (E 340)
2.2. Suspension (Système Adjuvant AS01E)
- Dioléoyl phosphatidylcholine (E 322)
- Cholestérol
- Chlorure de sodium
- Phosphate disodique anhydre (E 339)
- Dihydrogénophosphate de potassium (E 340)
- Eau pour préparations injectables
Indications
Arexvy est indiqué pour l'immunisation active en vue de la prévention des maladies des voies respiratoires inférieures causées par le virus respiratoire syncytial :
- chez les adultes âgés de 60 ans et plus.
- chez les adultes de 50 à 59 ans à risque accru de maladie à VRS.
Ce vaccin doit être utilisé conformément aux recommandations officielles.
Recommandation de la Haute Autorité de santé (HAS) du 27 juin 2024 : vaccination des personnes ≥ 75 ans, ou ≥ 65 ans et plus présentant une maladie respiratoire chronique pouvant s'aggraver en cas d'infection à VRS (particulièrement la BPCO) ou cardiaque (particulièrement l'insuffisance cardiaque). Cette recommandation est prise en compte par le carnet de vaccination numérique de MesVaccins.
Attention, ce vaccin n'est pas autorisé chez la femme enceinte.
Posologie
Arexvy est administré en une seule dose de 0,5 mL.
La nécessité d'une revaccination avec une dose supplémentaire n'a pas été établie (voir rubrique "Pharmacodynamie").
Cette vaccination est recommandée en période épidémique (soit de septembre à janvier) chez les personnes éligibles : ≥ 75 ans, ou ≥ 65 ans et plus présentant une maladie respiratoire chronique pouvant s'aggraver en cas d'infection à VRS (particulièrement la BPCO) ou cardiaque (particulièrement l'insuffisance cardiaque).
Population pédiatrique
La sécurité et l’efficacité d’Arexvy chez les enfants n’ont pas été établies. Aucune donnée n’est disponible.
Mode d'administration
Pour injection intramusculaire uniquement, de préférence dans le muscle deltoïde.
Pour les instructions concernant la reconstitution du médicament avant administration, voir rubrique "Manipulation".
Contre-indications
Hypersensibilité aux substances actives ou à l’un des excipients mentionnés à la rubrique "Composition".
Mises en garde et précautions d'emploi
Traçabilité
Afin d'améliorer la traçabilité des médicaments biologiques, le nom et le numéro de lot du produit administré doivent être clairement enregistrés.
Avant immunisation
Un traitement médical approprié doit être disponible immédiatement et une surveillance doit être effectuée au cas où une réaction anaphylactique surviendrait après l'administration du vaccin.
La vaccination doit être différée chez les sujets souffrant d'une maladie fébrile aiguë sévère. La présence d'une infection mineure, telle qu'un rhume, ne doit pas conduire au report de la vaccination.
Comme pour tout vaccin, une réponse immunitaire protectrice peut ne pas être obtenue chez tous les sujets vaccinés.
Des réactions liées à l'anxiété, y compris des réactions vasovagales (syncope), une hyperventilation ou des réactions liées au stress peuvent survenir en lien avec le processus de vaccination lui-même. Il est important que des mesures soient mises en place afin d'éviter des blessures en cas d'évanouissement.
Précautions d'emploi
Ne pas administrer le vaccin par voie intravasculaire ou intradermique. Aucune donnée n'est disponible sur l'administration sous-cutanée d’Arexvy.
Comme pour les autres injections intramusculaires, Arexvy doit être administré avec précaution chez les sujets atteints de thrombopénie ou d'un trouble de la coagulation car un saignement peut se produire chez ces sujets après une administration intramusculaire.
Traitements immunosuppresseurs systémiques et déficit immunitaire
Il n'existe pas de données de sécurité et d'immunogénicité d’Arexvy chez les sujets immunodéprimés. Les patients recevant un traitement immunosuppresseur ou souffrant d'un déficit immunitaire peuvent avoir une réponse immunitaire réduite à Arexvy.
Excipients
Ce médicament contient moins de 1 mmol (39 mg) de potassium par dose, c'est-à-dire qu’il est essentiellement « sans potassium ».
Ce médicament contient moins de 1 mmol (23 mg) de sodium par dose, c'est-à-dire qu’il est essentiellement « sans sodium ».
Interactions
Utilisation avec d'autres vaccins
Paragraphe ajouté le 26 novembre 2025 :
Arexvy peut être administré de manière concomitante avec un vaccin à ARNm contre la covid 19, un vaccin pneumococcique conjugué, un vaccin contre le zona (recombinant, avec adjuvant) ou avec un vaccin inactivé contre la grippe saisonnière (dose standard sans adjuvant, haute dose sans adjuvant, ou dose standard avec adjuvant).
Si Arexvy doit être administré en même temps qu'un autre vaccin injectable, les vaccins doivent toujours être administrés en des sites d'injection différents.
L'administration concomitante d’Arexvy avec d'autres vaccins que ceux listés ci-dessus n'a pas été étudiée.
D'une manière générale, on considère toutefois qu'on peut administrer deux vaccins non vivants le même jour ou à n'importe quel intervalle.
Fertilité
Il n'existe pas de donnée sur les effets d’Arexvy sur la fertilité humaine. Les études chez l’animal menées avec Arexvy ou avec le vaccin expérimental RSVPreF3 sans adjuvant n'indiquent pas d'effets délétères directs ou indirects en ce qui concerne la toxicité sur la reproduction. Voir rubrique "Autres informations / Données de sécurité préclinique").
Grossesse
Il n'existe pas de données sur l'utilisation d’Arexvy chez la femme enceinte. Après l'administration du vaccin expérimental RSVPreF3 sans adjuvant à 3 557 femmes enceintes dans un seul essai clinique, une augmentation des naissances prématurées a été observée par rapport au placebo. Actuellement, aucune conclusion ne peut être établie sur une relation de cause à effet entre l'administration de RSVPreF3 sans adjuvant et les naissances prématurées. Les résultats des études chez l’animal avec Arexvy ou avec le vaccin expérimental RSVPreF3 sans adjuvant n'indiquent pas d'effets délètères directs ou indirects en ce qui concerne la toxicité sur le développement et la reproduction (voir rubrique "Autres informations / Données de sécurité préclinique"). Arexvy n'est pas recommandé pendant la grossesse.
Allaitement
Il n'existe pas de données sur l’excrétion d’Arexvy dans le lait de la femme ou de la femelle chez l’animal. Arexvy n'est pas recommandé chez la femme qui allaite.
Effets indésirables
1. Résumé du profil de sécurité
Le profil de sécurité présenté dans le Tableau 1 est basé sur une analyse groupée des données générées dans deux études cliniques de phase III contrôlées par placebo (menées en Europe, en Amérique du Nord, en Asie et dans l'hémisphère Sud) chez des adultes âgés de ≥ 60 ans, et de 50 à 59 ans, ainsi que sur la période post-commercialisation.
Chez les participants à l'étude âgés de 60 ans et plus (plus de 12 000 adultes ont reçu une dose d’Arexvy et plus de 12 000 ont reçu un placebo, avec une période de suivi d’environ 12 mois), les effets indésirables les plus fréquemment rapportés étaient une douleur au site d'injection (61 %), une fatigue (34 %), une myalgie (29 %), une céphalée (28 %) et une arthralgie (18 %). Ces effets indésirables étaient généralement d'intensité légère ou modérée et disparaissaient quelques jours après la vaccination.
La plupart des autres effets indésirables ont été peu fréquents et rapportés de manière similaire entre les groupes de l'étude.
Chez les participants de l'étude âgés de 50 à 59 ans (769 participants, dont 386 participants présentant des pathologies chroniques prédéfinies et stables entraînant un risque accru de maladie à VRS), une incidence plus élevée de douleur au point d'injection (76 %), de fatigue (40 %), de myalgie (36 %), de céphalées (32 %) et d'arthralgie (23 %) a été observée, par rapport aux participants âgés de 60 ans et plus (381 participants) de la même étude. Cependant, la durée et la gravité de ces événements étaient comparables dans tous les groupes d'âge de l'étude.
Liste tabulée des effets indésirables
Les effets indésirables sont repris ci-dessous par classe de systèmes d’organes MedDRA et par fréquence.
- très fréquent (≥ 1/10) ;
- fréquent (≥ 1/100 à < 1/10) ;
- peu fréquent (≥ 1/1 000 à < 1/100) ;
- rare (≥ 1/10 000 à < 1/1 000) ;
- très rare (< 1/10 000) ;
- indéterminée (la fréquence ne peut être estimée à partir des données disponibles).
Effets indésirables observés lors des essais cliniques ainsi que les effets indésirables qui ont été signalés spontanément lors de l'utilisation d'Arexvy après la commercialisation dans le monde entier.
Affections hématologiques et du système lymphatique
- Peu fréquent : lymphadénopathie.
Affections du système immunitaire
- Peu fréquent : réactions d'hypersensibilité (telles que rash).
Affections du système nerveux
- Très fréquent : céphalée.
- Très rare : syndrome de Guillain-Barré.
Affections gastro-intestinales
- Peu fréquent : nausées, douleur abdominale, vomissements.
Affections musculosquelettiques et du tissu conjonctif
- Très fréquent : myalgie, arthralgie.
Troubles généraux et anomalies au site d'administration
- Très fréquent : douleur au site d’injection, érythème au site d'injection, fatigue.
- Fréquent : gonflement au site d'injection, fièvre, frissons.
- Peu fréquent : prurit au site d'injection, douleur, malaise.
- Indéterminée : nécrose au site d'injection 1.
Description des effets indésirables sélectionnées
Dans une étude observationnelle post-commercialisation menée aux États-Unis chez des personnes âgées de 65 ans et plus, une augmentation du risque de survenue d’un syndrome de Guillain-Barré (estimé à 7 cas supplémentaires par million de doses administrées) a été observée au cours des 42 jours suivant la vaccination avec Arexvy.
2. Effets sur l’aptitude à conduire des véhicules et à utiliser des machines
Aucune étude concernant les effets d’Arexvy sur l'aptitude à conduire des véhicules et à utiliser des machines n'a été réalisée.
Arexvy a une influence mineure sur l’aptitude à conduire des véhicules et à utiliser des machines. Certains effets mentionnés à la rubrique "Effets indésirables" (par ex. fatigue) peuvent temporairement affecter la capacité à conduire des véhicules ou à utiliser des machines.
3. Surdosage
Aucun cas de surdosage n’a été signalé dans les études cliniques.
4. Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : www.signalement-sante.gouv.fr.
Pharmacodynamie
Classe pharmacothérapeutique : Vaccins, autres vaccins viraux.
Code ATC : J07BX05.
Mécanisme d’action
En combinant l'antigène spécifique au VRS, protéine F en forme pré-fusion, avec un système adjuvant (AS01E), Arexvy est conçu pour renforcer les réponses immunitaires cellulaires spécifiques à l’antigène et par anticorps neutralisants chez des sujets possédant déjà une immunité contre le VRS. L'adjuvant AS01E facilite le recrutement et l'activation des cellules présentatrices d'antigènes transportant les antigènes du vaccin dans le ganglion lymphatique drainant, ce qui conduit à la production de lymphocytes T CD4+ spécifiques du RSVPreF3.
Efficacité
L'efficacité contre une MVRI associée au VRS, chez les adultes de 60 ans et plus, a été évaluée jusqu’à 3 saisons de VRS dans le cadre d'une étude clinique de phase III, randomisée, contrôlée par placebo et menée en simple aveugle (insu de l'observateur) dans 17 pays de l'hémisphère Nord et de l'hémisphère Sud.
La population principale utilisée pour l'analyse d'efficacité (référencée comme population exposée modifiée) incluait les adultes de 60 ans et plus qui ont reçu 1 dose d’Arexvy ou de placebo et n'ayant pas déclaré d’infection respiratoire aiguë [IRA] confirmée liée au VRS avant le 15ème jour suivant la vaccination.
Au total, 24 960 participants ont été randomisés de manière égale dans le groupe recevant 1 dose d’Arexvy (N = 12 466) ou dans le groupe placebo (N = 12 494) pendant la première saison. Avant la saison 2, les participants, qui ont reçu Arexvy pendant la première saison, ont été de nouveau randomisés pour recevoir un placebo (N= 4 991) ou une seconde dose d'Arexvy (N= 4 966). Les participants qui ont reçu un placebo avant la saison 1 ont reçu une seconde dose de placebo avant la saison 2. Les participants ont été suivis jusqu'à la fin de la troisième saison de VRS (période médiane de suivi de 30,6 mois).
L'âge médian des participants était de 69 ans (intervalle : 59 à 102 ans), dont environ 74 % avaient plus de 65 ans, environ 44 % plus de 70 ans et environ 8 % plus de 80 ans. Environ 52 % des participants étaient des femmes.
À l'inclusion, 39,3 % des participants présentaient au moins une comorbidité d’intérêt ; 19,7 % des participants avaient une maladie cardiorespiratoire sous-jacente (BPCO, asthme, toute maladie respiratoire/pulmonaire chronique ou insuffisance cardiaque chronique) et 25,8 % des participants avaient une maladie endocrino-métabolique (diabète, maladie hépatique ou rénale avancée).
Les cas confirmés de VRS ont été déterminés par une réaction quantitative en chaîne par polymérase à transcription inverse (qRT-PCR) sur un prélèvement nasopharyngé. La MVRI a été définie sur la base des critères suivants : le participant doit avoir présenté au moins deux symptômes/signes respiratoires des voies inférieures, dont au moins un signe respiratoire des voies inférieures pendant au moins 24 heures, ou avoir présenté au moins trois symptômes respiratoires des voies inférieures pendant au moins 24 heures. Les symptômes respiratoires des voies inférieures comprenaient : expectorations nouvelles ou accrues, toux nouvelle ou accrue, dyspnée nouvelle ou accrue (souffle court). Les signes respiratoires des voies inférieures comprenaient : respiration sifflante nouvelle ou accrue, crépitations/sibilances, fréquence respiratoire ≥ 20 respirations/min, saturation en oxygène faible ou diminuée (saturation en O2 < 95 % ou ≤ 90 % si la valeur d'inclusion était < 95 %) ou besoin de supplémentation en oxygène.
Efficacité contre la MVRI associée au VRS pendant la première saison de VRS (analyse confirmatoire)
L'objectif principal était de démontrer l'efficacité dans la prévention d'un premier épisode confirmé de MVRI liée au VRS-A et/ou B au cours de la première saison de VRS.
L'efficacité du vaccin, globale et par sous-groupe, est présentée dans le tableau 1.
L'efficacité d’Arexvy dans la prévention du premier épisode de MVRI associée au VRS, apparue à partir du 15ème jour après la vaccination, par rapport au placebo, était de 82,6 % (intervalle de confiance à 96,95 % de 57,9 % à 94,1 %) chez les participants âgés de 60 ans et plus. L'efficacité du vaccin contre la MVRI due au VRS a été observée pendant la période médiane de suivi de 6,7 mois.
L'efficacité du vaccin contre la MVRI associée au VRS-A et la MVRI associée au VRS-B était respectivement de 84,6 % (IC à 95 % [32,1 à 98,3]) et de 80,9 % (IC à 95 % [49,4 à 94,3]).
Tableau 1 - Analyse d'efficacité pendant la première saison de VRS (analyse confirmatoire) : premier épisode de MVRI associée au VRS dans la population globale, par âge et par sous-groupe de comorbidité (population exposée modifiée).


b Objectif confirmatoire avec un critère de réussite prédéfini de la limite inférieure de l'IC bilatéral supérieure à 20 % pour l'efficacité du vaccin ;
N = nombre de participants inclus dans chaque groupe ;
n = Nombre de participants ayant eu une première MVRI confirmée liée au VRS à partir du 15ème jour après la vaccination.
L'efficacité du vaccin dans le sous-groupe des participants âgés de 80 ans et plus (1 016 participants dans le groupe Arexvy vs 1 028 participants dans le groupe placebo) ne peut être estimée de manière fiable en raison du faible nombre de cas totaux observés (5 cas).
Parmi les 18 cas de MVRI due au VRS présentant au moins 2 signes des voies respiratoires inférieures ou empêchant les activités quotidiennes, 4 cas présentaient une MVRI sévère due au VRS, nécessitant une supplémentation en oxygène dans le groupe placebo contre aucun cas dans le groupe Arexvy.
Efficacité contre la MVRI associée au VRS pendant 2 saisons de VRS et pendant 3 saisons de VRS
Les participants âgés de 60 ans et plus ayant reçu une dose d'Arexvy ou de placebo ont été suivis pendant 3 saisons de VRS (jusqu'à la fin de la deuxième et troisième saisons dans l'Hémisphère Nord) avec une période médiane de suivi de 17,8 mois pendant 2 saisons de VRS et de 30,6 mois pendant 3 saisons de VRS. L’efficacité du vaccin contre la MVRI associée au VRS pendant 2 saisons de VRS était de 67,2% (IC à 97,5% [48,2 ; 80,0]) et pendant 3 saisons de VRS était de 62,9% (IC à 97,5% [46,7 , 74,8]).
L'efficacité du vaccin contre la MVRI associée au VRS-A et contre la MVRI associée au VRS-B pendant 3 saisons de VRS était respectivement de 69,8 % (IC à 97,5 % [42,2, 85,7]) et de 58,6 % (IC à 97,5 % [35,9, 74,1]).
L’efficacité du vaccin contre la MVRI associée au VRS était similaire chez les participants avec au moins une comorbidité d’intérêt.
Une seconde dose de vaccin administrée 12 mois après la première dose n’a pas conféré de bénéfice supplémentaire en termes d’efficacité.
Immunogénicité chez les adultes âgés de 50 à 59 ans à risque accru de maladie à VRS
La non-infériorité de la réponse immunitaire à Arexvy chez les adultes âgés de 50 à 59 ans par rapport aux adultes âgés de 60 ans et plus, pour lesquels l'efficacité du vaccin contre la MVRI associée au VRS a été démontrée, a été évaluée dans le cadre d'une étude de phase III, en aveugle, randomisée et contrôlée par placebo.
La cohorte 1 était composée de participants âgés de 50 à 59 ans répartis en deux sous-cohortes (Adultes-AIR et Adultes-non-AIR) en fonction de leurs antécédents médicaux. La sous-cohorte Adultes-AIR (adultes à risque accru) était composée de participants présentant des pathologies chroniques prédéfinies et stables, entraînant un risque accru de maladie à VRS (Arexvy, N= 386 ; placebo, N= 191), telles que maladie pulmonaire chronique, maladie cardiovasculaire chronique, diabète, maladie rénale ou hépatique chronique. La sous-cohorte Adultes-non-AIR était composée de participants ne souffrant pas de pathologies chroniques prédéfinies et stables (Arexvy, N= 383 ; placebo, N= 192). La cohorte 2 (OA ; adultes âgés) était composée de participants âgés de 60 ans et plus (Arexvy, N= 381).
Les objectifs primaires d'immunogénicité étaient de démontrer la non-infériorité de la réponse immunitaire humorale (en termes de titres de neutralisation viraledu VRS-A et du VRS-B) après l'administration d'Arexvy, un mois post-vaccination chez les participants âgés de 50 à 59 ans avec ou sans pathologie chronique prédéfinie et stable, entraînant un risque accru de maladie à VRS, par rapport aux participants âgés de 60 ans et plus.
Table 2 - Résumé des valeurs de MGT ajustées et de SRR, des ratios de MGT ajustés et des différences de SRR en termes de titres de neutralisation virale du VRS-A et du VRS-B (ED60) chez les adultes âgés de 60 ans et plus (OA) par rapport aux adultes âgés de 50 à 59 ans avec (Adultes-AIR) et sans (Adultes-non-AIR) pathologies chroniques prédéfinies et stablesa, conduisant à un risque accru de maladie à VRS – analyse per protocole.
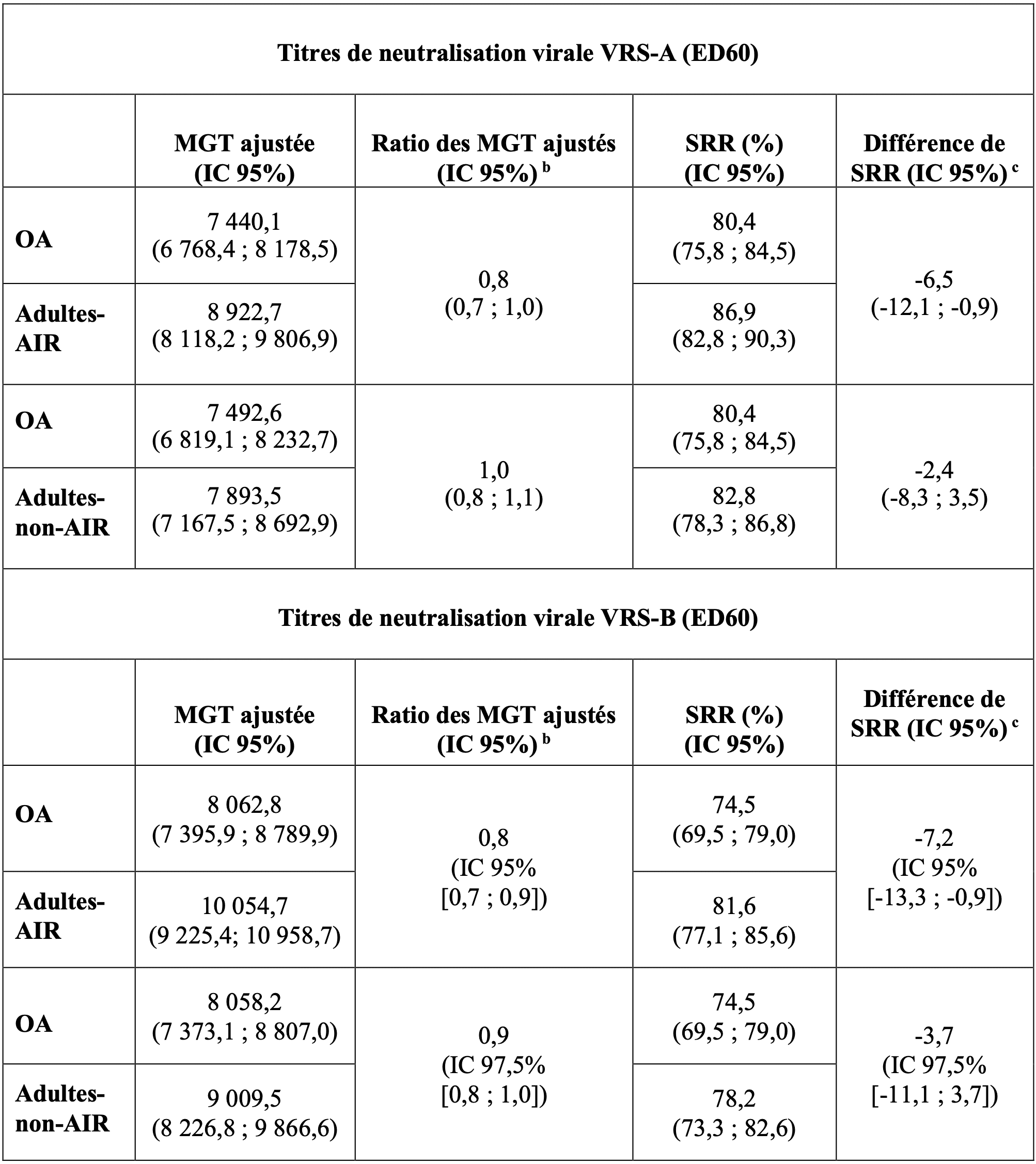
chronique, diabète, maladie rénale ou hépatique chronique.
b,c. Les critères préspécifiés de non-infériorité des réponses immunitaires ont été définis comme les limites supérieures (LS) de l'IC bilatéral à 95 % ou 97,5 % sur les rapports MGT ajustés (OA par rapport à Adultes-AIR ou Adultes-non-AIR) ≤ 1,5 et la LS de l'IC bilatéral à 95 % ou 97,5% sur la différence de SRR (OA moins Adultes-AIR ou Adultes-non-AIR) ≤ 10% chez les participants âgés de 60 ans et plus (OA) par rapport aux participants âgés de 50 à 59 ans avec (Adultes-AIR) ou sans (Adultes-non-AIR) pathologies chroniques prédéfinies et stables, conduisant à un risque accru de maladie à VRS.
ED60 : Dilution estimée 60 ; IC = Intervalle de confiance ; MGT = Moyenne géométrique des titres ; SRR = taux de séroréponse
Les critères de non-infériorité des réponses immunitaires pour les titres de neutralisation virale du VRS-A et du VRS-B ont été atteints. L'efficacité d'Arexvy, chez les adultes âgés de 50 à 59 ans présentant un risque accru de maladie à VRS, peut être établie en comparant la réponse immunitaire chez les adultes âgés de 50 à 59 ans à la réponse immunitaire chez les adultes âgés de 60 ans et plus chez lesquels l'efficacité du vaccin a été démontrée.
Population pédiatrique
L’Agence Européenne des Médicaments a différé l’obligation de soumettre les résultats d’études réalisées avec Arexvy dans un ou plusieurs sous-groupes de la population pédiatrique dans la prévention de la maladie des voies respiratoires inférieures causée par le virus respiratoire syncytial (voir rubrique 4.2 pour les informations concernant l'usage pédiatrique).
Conservation
3 ans.
Après reconstitution :
La stabilité physicochimique a été démontrée pendant 4 heures à une température comprise entre 2 °C et 8 °C ou à température ambiante ne dépassant pas 25 °C.
D’un point de vue microbiologique, le produit doit être utilisé immédiatement. S'il n'est pas utilisé immédiatement, les durées et conditions de conservation avant utilisation sont de la responsabilité de l'utilisateur ; elles ne doivent pas dépasser 4 heures.
A conserver au réfrigérateur (entre 2°C et 8°C).
Ne pas congeler.
A conserver dans l’emballage extérieur d’origine à l’abri de la lumière.
Manipulation
La poudre et la suspension doivent être reconstituées avant administration.
 |
La poudre et la suspension doivent être inspectées visuellement pour mettre en évidence la présence de particules étrangères et/ou un changement d'apparence. Dans l'un ou l'autre de ces cas, ne pas reconstituer le vaccin.
En l’absence d’études de compatibilité, ce médicament ne doit pas être mélangé avec d’autres médicaments.
Comment préparer Arexvy
Arexvy doit être reconstitué avant administration.
- Retirer tout le contenu du flacon contenant la suspension dans une seringue.
- Ajouter tout le contenu de la seringue dans le flacon contenant la poudre.
- Agiter doucement jusqu'à ce que la poudre soit totalement dissoute.
Le vaccin reconstitué est un liquide opalescent, incolore à brun pâle.
Le vaccin reconstitué doit être inspecté visuellement pour mettre en évidence la présence de particules étrangères et/ou un changement d'apparence. Dans l'un ou l'autre de ces cas, ne pas administrer le vaccin.
La stabilité physicochimique a été démontrée pendant 4 heures à une température comprise entre 2 °C et 8 °C ou à température ambiante ne dépassant pas 25 °C.
D’un point de vue microbiologique, le produit doit être utilisé immédiatement. S'il n'est pas utilisé immédiatement, les durées et conditions de conservation avant utilisation sont de la responsabilité de l'utilisateur ; elles ne doivent pas dépasser 4 heures.
Avant administration :
- Retirer 0,5 mL du vaccin reconstitué dans la seringue.
- Changer l'aiguille de façon à utiliser une nouvelle aiguille.
Administrer le vaccin par voie intramusculaire
Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
L'administration du vaccin doit être consignée par le médecin sur le carnet de vaccination ou de santé avec le numéro de lot. Il peut aussi être inscrit par le patient ou son médecin sur un carnet de vaccination numérique (version grand public ou professionnelle).
Autres informations
Autorisé dans l'Union européenne depuis le 16 juin 2023.
Données de sécurité préclinique
Les données non cliniques issues des études conventionnelles de toxicité après administrations répétées n’ont pas révélé de risque particulier pour l’homme.
Les études sur la reproduction et le développement réalisées chez le lapin avec Arexvy ou avec le vaccin RSVPreF3 sans adjuvant n'ont pas révélé d'effets liés au vaccin sur la fertilité des femelles, la grossesse, ou le développement embryofœtal ou le développement de la progéniture.